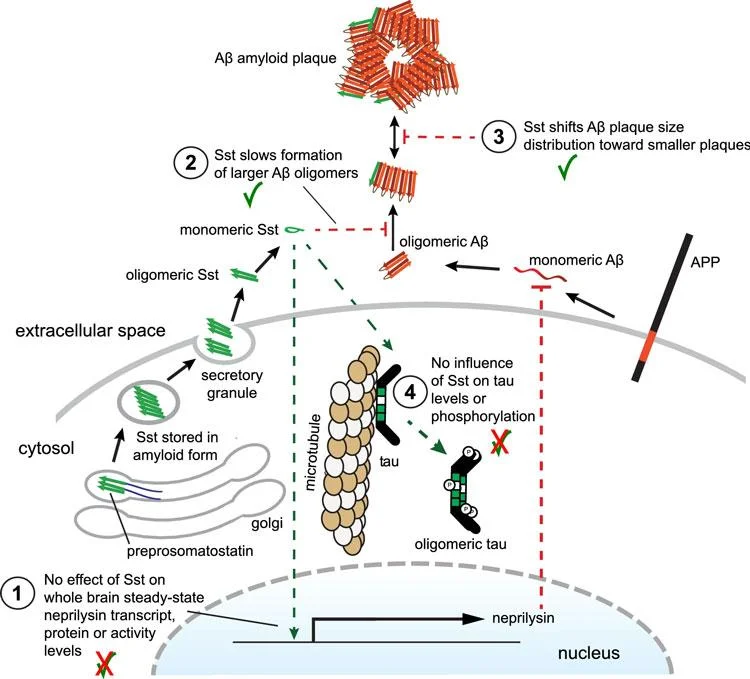
These observations suggested a relationship between somatostatin and amyloid beta aggregation, but researchers did not know what it was.
Then, in the early 2000s, a team from Japan published research describing that somatostatin drives production of an enzyme called neprilysin that can degrade amyloid beta. This finding suggested that if somatostatin was decreased, the levels of neprilysin would decrease – without neprilysin to degrade amyloid beta, the plaques would continue to grow, and Alzheimer’s disease would progress.
This understanding of the role of somatostatin is where the field stood for more than a decade.
The impact of somatostatin
Several years ago, Schmitt-Ulms and his team investigated amyloid beta in its monomer and oligomer forms, specifically looking for molecules in the brain that would bind to the toxic oligomeric forms. They found that, of all proteins in the brain, somatostatin was the smallest to bind to amyloid beta – and the most selective, as it only interacted with the oligomers.
They then observed that somatostatin blocked the formation of oligomers, with higher concentrations of somatostatin having a greater effect. This earlier study provided the first evidence that somatostatin interacts directly with the oligomeric amyloid beta that are known to be the earliest stages of Alzheimer’s disease, providing an alternative perspective on the impact of somatostatin on the disease.
“We didn’t actually intend to work on somatostatin, but these results made us very curious about what would happen if somatostatin was absent in an animal model that was genetically engineered to develop amyloid beta aggregations,” Schmitt-Ulms explains.
A striking finding
To answer this question, the research team crossed a somatostatin-deficient mouse line with an amyloidosis mouse model. They found that when somatostatin was absent, there were more amyloid beta aggregates, consistent with what is seen in Alzheimer’s disease in humans.
Remarkably, the result did not seem to be caused by an effect of somatostatin on neprilysin levels, as there were no differences – a contradiction to the long-held understanding of the mechanism through which somatostatin impacts Alzheimer’s disease. Instead, the data suggested that somatostatin blocks oligomer formation independent of neprilysin by interacting with amyloid beta.
“There isn’t a lot of literature that explains how the environment in the brain can promote or inhibit amyloid beta forming into oligomers, so this is a nice vignette that shows that one molecule – somatostatin – seems to influence that first small oligomeric aggregation step,” Schmitt-Ulms says.
“And if you get fewer of these oligomeric forms, then you get fewer of the small clumps, which are the next step of amyloid beta aggregation – so the process made sense, and it was a striking finding.”
Prompting discussions in the scientific community